The Federal Circuit’s September 23, 2025 decision in Bayer Pharma Aktiengesellschaft v. Mylan Pharmaceuticals Inc., No. 23-2434, delivers a sharp reminder that post-hoc validation, even in the form of successful clinical trial results, does not automatically translate into patentability. The opinion clarifies the limits of method-of-treatment claims that attempt to rely on clinical proof language to distinguish otherwise anticipated dosing regimens, while also providing important guidance on claim construction in combination-therapy claims.
The case arose from inter partes review proceedings challenging U.S. Patent No. 10,828,310, which describes the results of Bayer’s COMPASS Phase III clinical trial evaluating low-dose rivaroxaban in combination with aspirin to reduce major adverse cardiovascular events in patients with coronary artery disease (CAD) and peripheral artery disease (PAD). While the clinical trial itself was significant, the Federal Circuit emphasized that patentability must turn on what the claims require a practitioner to do, not on what later evidence proves about the outcomes.
The court ultimately affirmed the PTAB’s unpatentability determinations for claims 1–4, vacated the unpatentability determinations for claims 5–8 based on an erroneous claim construction, and remanded for further proceedings. Along the way, the opinion addressed four recurring issues in pharmaceutical patent litigation: (1) the role of “clinically proven effective” language, (2) construction of combination-product claims, (3) motivation to combine prior art clinical disclosures, and (4) the limits of unexpected-results evidence.
The COMPASS Claims and the Prior Art
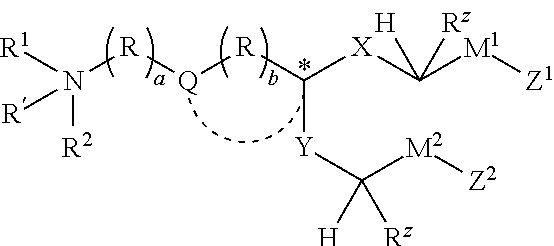
The ’310 patent claims methods of reducing the risk of myocardial infarction, stroke, or cardiovascular death by administering rivaroxaban at 2.5 mg twice daily together with aspirin at 75–100 mg daily. Claims 1–4 broadly recite administering those agents at the specified dosages, while claims 5–8 add a requirement that the method involve “once daily administration of a first product comprising rivaroxaban and aspirin” and “a second product comprising rivaroxaban.”
The IPR petitions relied primarily on two references. The first was a 2016 journal article by Foley summarizing the then-ongoing COMPASS trial, including the exact dosing regimen later claimed, but without disclosing the trial’s results. The second was a 2014 article by Plosker describing the ATLAS ACS 2-TIMI 51 trial, which disclosed similar low-dose rivaroxaban plus aspirin regimens in cardiovascular patients.
The PTAB concluded that claims 1–2 were anticipated by Foley and that claims 1–8 were obvious over Foley alone or in combination with Plosker. Bayer appealed.
“Clinically Proven Effective” and the Functional Relationship Test
A central dispute on appeal concerned the claim phrase “clinically proven effective.” Bayer argued that this language was limiting and required proof of efficacy through clinical trials, which Foley did not disclose. The Board had concluded that the phrase was non-limiting, or alternatively, inherently anticipated.
The Federal Circuit sidestepped that debate entirely. Instead, it held that even if “clinically proven effective” were treated as a limiting element, it could not confer patentability because it lacked a new and unobvious functional relationship with the claimed method. Drawing heavily on King Pharmaceuticals, Inc. v. Eon Labs, Inc., 616 F.3d 1267 (Fed. Cir. 2010), the court explained that an otherwise anticipated method cannot be rescued by adding a limitation that merely describes information about the method or its later validation.
As the court put it, proof that a dosing regimen worked in a clinical trial “in no way transforms the process of taking the drugs” at the claimed doses and frequencies. The actual steps performed by the physician or patient remain unchanged, regardless of whether the regimen has been clinically validated. Allowing patentees to “claw back” known treatment methods from the public domain simply by pointing to later-obtained clinical success would undermine fundamental limits on patent scope.
Charles Gideon Korrell notes that this portion of the opinion is particularly important for pharmaceutical lifecycle management strategies that rely on clinical trial milestones rather than changes to dosing, formulation, or administration. The court’s analysis reinforces that validation alone is not innovation, at least for purposes of patentability.
Distinguishing Allergan and Open-Ended Claim Drafting
Bayer argued that Allergan Sales, LLC v. Sandoz, Inc., 935 F.3d 1370 (Fed. Cir. 2019), compelled a different result. In Allergan, the court held that certain “wherein” clauses specifying minimum safety and efficacy thresholds were limiting and material to patentability.
The Federal Circuit rejected the comparison. Unlike the open-ended composition claims in Allergan, which could encompass a wide range of formulations meeting the recited concentration requirements, the Bayer claims already fixed the precise dosages of rivaroxaban and aspirin. The “clinically proven effective” language did not further narrow the universe of covered methods. As Charles Gideon Korrell observes, the opinion underscores a drafting lesson: functional or results-based language is far more likely to matter when it genuinely constrains an otherwise open claim scope.
Claim Construction of the “First Product”
While Bayer lost on claims 1–4, it prevailed on an important claim construction issue affecting claims 5–8. The PTAB had construed “a first product comprising rivaroxaban and aspirin” to encompass administration of the two drugs as separate dosage forms, whether administered simultaneously or sequentially.
The Federal Circuit disagreed. Focusing on the plain claim language, the court held that “a first product comprising rivaroxaban and aspirin” requires a single dosage form that includes both active ingredients. Interpreting the term to cover separate pills would render the “first product” language meaningless and collapse the distinction the claims draw between combination and non-combination products.
The court also found support in the specification, which explicitly distinguishes between “separate dosage forms” and “a combination dosage form containing both rivaroxaban and aspirin.” The Board’s broader construction improperly conflated these concepts. Because the PTAB had not analyzed obviousness under the correct construction, the Federal Circuit vacated the unpatentability determinations for claims 5–8 and remanded.
Charles Gideon Korrell believes this portion of the decision highlights how seemingly modest wording choices can drive materially different outcomes in IPR proceedings, particularly for combination-product claims in the pharmaceutical space.
Motivation to Combine and Reasonable Expectation of Success
Bayer also challenged the Board’s obviousness analysis for dependent claims reciting specific aspirin dosages (75 mg or 81 mg). The Federal Circuit was unpersuaded. The Board had expressly found that these dosages reflected globally available aspirin strengths and were consistent with the dosage ranges disclosed in Plosker.
The court reiterated that motivation to combine and reasonable expectation of success are factual determinations reviewed for substantial evidence. Here, the record supported the Board’s conclusion that a skilled artisan would have had reason to substitute commonly used aspirin dosages with an expectation of success, particularly given that both references addressed reducing cardiovascular risk.
Unexpected Results and the Nexus Requirement
Finally, Bayer argued that the clinical proof of efficacy demonstrated by the COMPASS trial constituted unexpected results supporting nonobviousness. The Federal Circuit rejected this argument on nexus grounds.
The Board had found, and the court agreed, that Bayer’s evidence of unexpected results was tied exclusively to the “clinically proven effective” limitation. Because that limitation was functionally unrelated to the claimed method steps and could not supply patentability, the asserted unexpected results lacked the required nexus to the merits of the claimed invention.
As Charles Gideon Korrell notes, this analysis reinforces the principle that secondary considerations must be anchored to what is both claimed and novel. Evidence of success attributable to validation, rather than to a structural or functional distinction over the prior art, will not carry the day.
Takeaways
The Bayer decision offers several practical lessons. First, method-of-treatment claims cannot rely on post-hoc clinical validation to rescue known dosing regimens from anticipation or obviousness. Second, careful claim drafting around combination products matters, and courts will enforce distinctions between single-dosage-form products and loose combinations of separate components. Third, secondary considerations must be tied to claim features that actually change how the method operates.
In short, the Federal Circuit reaffirmed that patent law rewards innovation in what practitioners do, not merely confirmation that what they were already doing turns out to work.